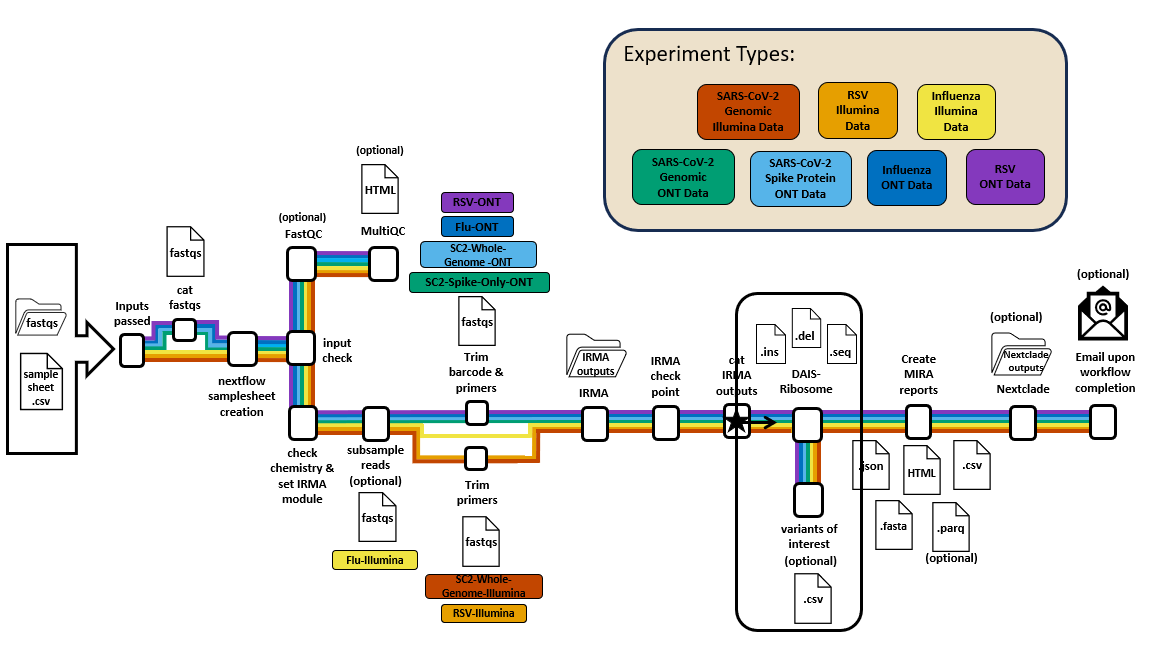
Content developed by Kristine Lacek
Genetic Markers in Influenza
When comparing amino acid sequences across influenza isolates, certain mutations carry particular biological significance. These mutations — known as genetic markers — can affect how the virus behaves in important ways:
- Antiviral resistance — Mutations that reduce the effectiveness of antiviral drugs (e.g., oseltamivir/Tamiflu)
- Mammalian adaptation — Mutations in avian influenza viruses that enhance replication in mammalian hosts, raising pandemic concern
- Antigenic sites — Changes at positions on surface proteins (HA, NA) that affect immune recognition and vaccine effectiveness
MIRA Flags Genetic Markers Automatically
MIRA includes built-in detection of known genetic markers during assembly. When a marker is identified in a sample, it is flagged in the output reports for analyst review.
Key Markers
| Gene | Marker | Significance |
|---|---|---|
| H1 NA | H275Y | Only clinically proven site for oseltamivir (neuraminidase inhibitor) resistance in H1N1 |
| H5N1 PB2 | E627K | Enhances viral replication in mammalian cells — a key indicator of mammalian adaptation in avian influenza |
How to Interpret Marker Flags
- Check the MIRA summary reports — flagged markers appear in the CSV and HTML output files under the genetic markers section
- Cross-reference with phenotypic data — if antiviral resistance is flagged, confirm with laboratory susceptibility testing when possible
- Consider the context — a mammalian adaptation marker in a human H5N1 isolate has different implications than the same marker in a poultry sample
- Review mixed populations — MIRA’s variant calling may detect a marker at sub-consensus frequency, indicating a mixed viral population where only some viruses carry the mutation
MIRA’s Find Positions of Interest Tool
The find_positions_of_int workflow is a tool that runs (or reruns) DAIS-ribosome and the find_positions_of_interest module. The module returns the codon and amino acid information at positions of interest that the user provides.
To get started, you’ll need to provide three inputs:
- DAIS-ribosome input — a concatenated FASTA file
- Reference table — reference genome sequences processed through DAIS-ribosome
- Positions of interest table — a list of mutations/positions you want to check
Once complete, you’ll get a CSV file listing all positions in your input sequences that match the positions of interest you specified.
 find_positions_of_int workflow
find_positions_of_int workflow
The DAIS-ribosome Input
The input file for this workflow is a concatenated FASTA file with assembled sequences from all of your samples. If you’ve run the MIRA pipeline, you’ll find this input file named DAIS_ribosome_input.fasta in the dais-ribosome folder within the aggregate outputs.
The Reference Table
The reference table can be made by running your reference genomes through DAIS-ribosome and selecting the columns shown below. The file needs to be tab delimited. An example using Flu genomes:
isolate_id isolate_name subtype passage_history nt_id ctype reference_id protein aa_aln cds_aln
EPI_ISL_25690 A/common magpie/Hong Kong/5052/2007 A / H5N1 2b14fd2e8f738834298e9099f00e59d020ffc552 A_HA_H5 VT1203 HA-signal .....LLFAIVSLVKS ...............CTTCTTTTTGCAATAGTCAGCCTTGTTAAAAGC
EPI_ISL_140 A/Hong Kong/1073/99 A / H9N2 a591bc9ad3a54f705940ad8483684cfc278c742c A_HA_H9 BGD0994 HA-signal METISLITILLVVTASNA ATGGAAACAATATCACTAATAACTATACTACTAGTAGTAACAGCAAGCAATGCA
The Positions of Interest Table
The positions of interest table should be structured as shown below. These should be positions you are interested in checking across your samples. The file must be tab delimited.
subtype protein position mutation_of_int phenotypic_consensus
A / H1N1 HA 7 H inference description
A / H1N1 HA 8 Q inference description
A / H1N1 HA 94 N inference description
A / H1N1 HA 121 N inference description
B HA 94 N inference description
B HA 121 N inference description
Input Parameters
| Flag | Description |
|---|---|
profile |
singularity, docker, local, sge, slurm. Use docker or singularity for containerized runs. Use local for running on a local computer. |
input |
<FILE_PATH>/DAIS_ribosome_input.fasta — the concatenated FASTA file described above. Full file path required. |
outdir |
The file path where the output directory will be written. Full file path required. |
positions_of_interest |
<FILE_PATH>/positions_of_interest.txt — the positions table described above. Full file path required. |
reference_seq_table |
<FILE_PATH>/reference_table.txt — the reference table described above. Full file path required. |
dais_module |
The DAIS module used by DAIS-ribosome. Options: INFLUENZA, BETACORONAVIRUS, RSV |
Optional parameters (can be omitted to use defaults):
| Flag | Description |
|---|---|
process_q |
(required for HPC profile) Name of the processing queue for job submission. |
email |
(optional) Email address to receive a summary upon completion. |
Running the Workflow
nextflow run ./main.nf \
-profile <profile> \
--input <RUN_PATH>/samplesheet.csv \
--outdir <OUTDIR> \
--runpath <RUN_PATH> \
--e Find-Positions-Of-Interest \
--positions_of_interest <filepath>/muts_of_int_table.txt \
--reference_seq_table <filepath>/ref_table.txt \
--dais_module <DAIS_MODULE> \
--process_q <QUEUE_NAME> \
--email <EMAIL_ADDRESS>
Output
The output is a CSV file listing each position of interest found in your samples, along with the sample and reference codons and amino acids:
sample,reference_strain,gisaid_accession,ctype,dais_reference,protein,sample_codon,reference_codon,aa_mutation,phenotypic_consequence
s3_6,A/California/07/2009,EPI_ISL_227813,A_NA_N1,CALI07,NA,GAA,GAA,E:119:E,
s3_6,A/California/07/2009,EPI_ISL_227813,A_NA_N1,CALI07,NA,GGC,GGC,G:197:G,
s3_6,A/California/07/2009,EPI_ISL_227813,A_NA_N1,CALI07,NA,ATA,ATA,I:223:I,
s3_6,A/California/07/2009,EPI_ISL_227813,A_NA_N1,CALI07,NA,CAC,CAC,H:275:H,
s3_6,A/California/07/2009,EPI_ISL_227813,A_NA_N1,CALI07,NA,TGC,TGC,C:292:C,
s3_6,A/California/07/2009,EPI_ISL_227813,A_NA_N1,CALI07,NA,GAT,GAT,D:294:D,
s1_6,A/Hong Kong/4801/2014,EPI_ISL_233740,A_NA_N2,HK4801,NA,GAA,GAA,E:119:E,
s1_3,A/California/07/2009,EPI_ISL_227813,A_PA,HK4801,PA,ATA,ATA,I:38:I,
s3_3,A/California/07/2009,EPI_ISL_227813,A_PA,HK4801,PA,ATA,ATT,I:38:I,
s4_3,A/California/07/2009,EPI_ISL_227813,A_PA,HK4801,PA,ATA,NNN,I:38:X,amino acid information missing
The aa_mutation column uses the format Reference:Position:Sample — for example, H:275:H means the reference has Histidine (H) at position 275, and the sample also has Histidine (no mutation). A result like I:38:X with the note “amino acid information missing” indicates the sample codon could not be resolved (e.g., NNN).