Content developed by Kristine Lacek and Ben Rambo-Martin
Module Objectives
- Understand NGS file formats
- Manipulate files with Bash
- Write “wrapper” scripts
- Move FASTQs through MIRA-NF on the command line
- Understand assembly and annotation files (
.sam,.bam,.vcf) - View and interpret sequence alignments
- Use samtools and Bash one-liners to manipulate NGS files
- Install and use IGV to view BAM files
NGS Review
NGS (Next-Generation Sequencing) is a high-throughput DNA/RNA sequencing technology that builds on earlier methods such as Sanger sequencing (low throughput, one fragment at a time).
| Platform | Read type |
|---|---|
| Illumina | Paired-end short reads (150–300 bp) |
| Nanopore / PacBio | Long reads (1 000+ bp), single-end |
Fragmented reads come off the sequencer unordered and must be assembled to create a consensus genome.
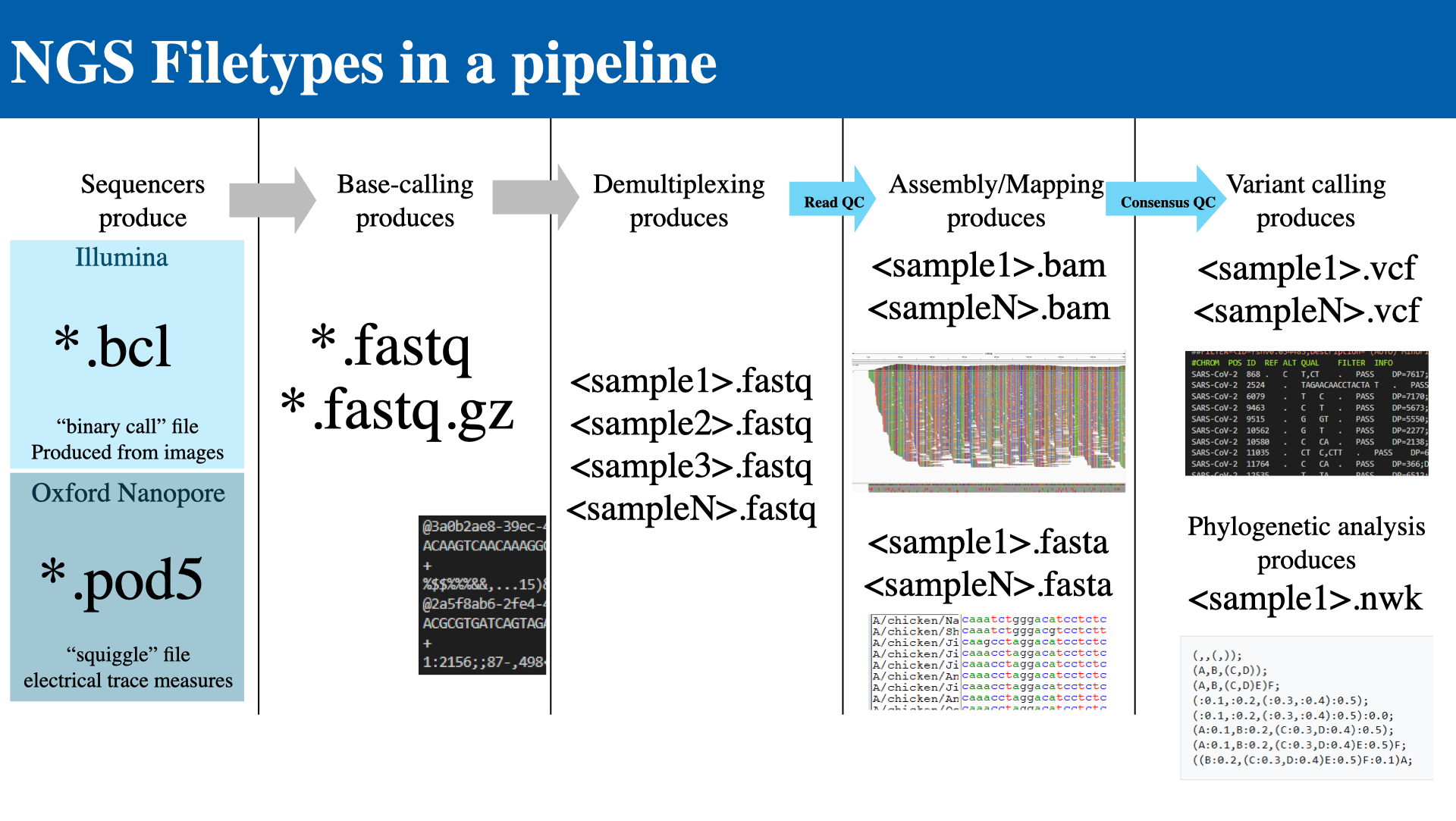
Standard NGS File Types
NGS Filetypes in a Pipeline
The following diagram shows how files flow through a typical NGS pipeline:
- Sequencers produce raw data
- Illumina →
.bcl(binary call files, produced from images) - Oxford Nanopore →
.pod5(electrical trace measurements)
- Illumina →
- Base-calling produces →
.fastq/.fastq.gz - Demultiplexing produces → per-sample
.fastqfiles- Each FASTQ record has four lines: Identifier, Sequence, +, Quality scores
- Assembly / Mapping produces →
.bamand.fastaper sample (with Read QC) - Variant calling produces →
.vcfper sample (with Consensus QC) - Phylogenetic analysis produces →
.nwk(Newick tree format)
Other Common File Types
| Format | Description | Common use |
|---|---|---|
| HTML | HyperText Markup Language — creates web page structure using tags | Reports |
| YAML | Human-readable data serialization using indentation | Configurations |
| MD (Markdown) | Lightweight markup for formatting plain text | Documentation, wikis |
| JSON | Structured data using key-value pairs | Figures, configs |
| XML | Structured data using custom tags and hierarchical elements | NCBI submissions |
BCL and FAST5 Files
BCL Files (Illumina)
- Raw output from Illumina sequencers, generated directly by the instrument during a run
- Contain base calls and quality scores — one file per cycle, per lane
- Not human-readable; must be converted before analysis
- Converted to FASTQ using tools like
bcl2fastq
FAST5 Files (Oxford Nanopore)
- Raw signal-level data — electrical current measurements from nanopores
- Contain rich metadata: timing, channel information, and raw signals
- Used for base-calling and re-analysis (allows re-base-calling as algorithms improve)
- Converted to FASTQ using tools like Guppy or Dorado
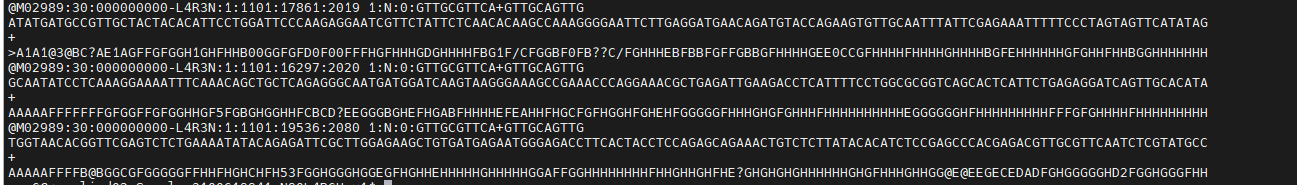
FASTQ Files
FASTQ is the standard text-based format for sequencing reads, storing both sequence and quality information.
Each read occupies four lines:
| Line | Content |
|---|---|
| 1 | Read identifier (starts with @) |
| 2 | Nucleotide sequence |
| 3 | Separator (+) |
| 4 | Quality scores (ASCII characters) |
One quality score per base — the quality line is always the same length as the sequence line. Files are commonly compressed as .fastq.gz.
Phred Quality Scores
Phred quality scores measure base-calling confidence on a logarithmic scale:
Q = −10 × log10(Perror)
Scores are encoded as ASCII characters and stored on line 4 of the FASTQ record.
| Score | Error rate | Accuracy |
|---|---|---|
| Q20 | 1 in 100 (1%) | 99% |
| Q30 | 1 in 1 000 (0.1%) | 99.9% |
| Q40 | 1 in 10 000 (0.01%) | 99.99% |
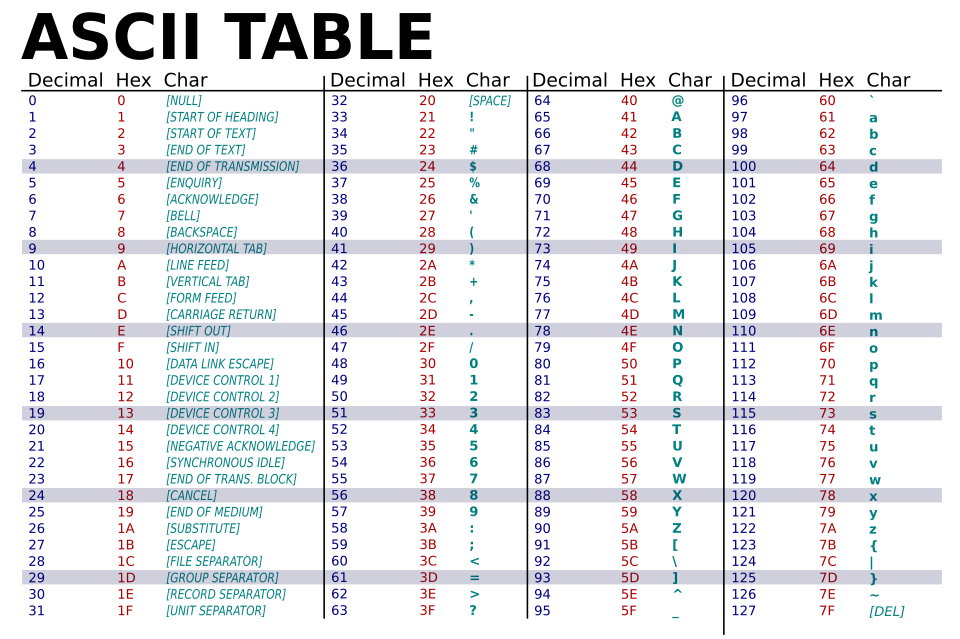
Figure credit: https://commons.wikimedia.org/wiki/File:ASCII-Table-wide.svg
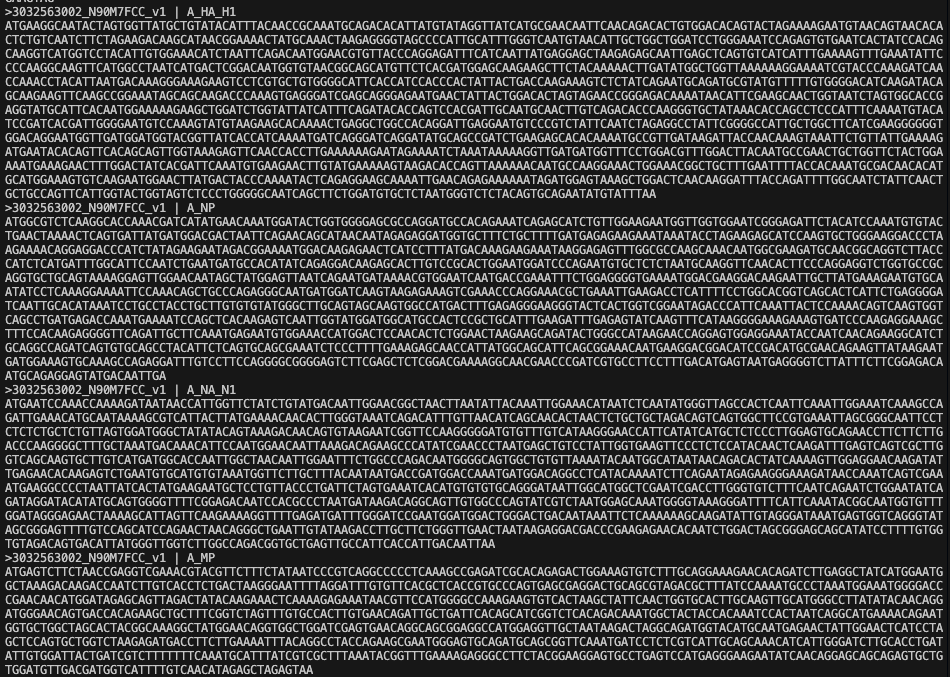
FASTA Files
FASTA is a simple text-based sequence format for DNA, RNA, or protein sequences.
- Header line begins with
> - Sequence lines follow the header
- A file with multiple sequences is called a multifasta
- Contains no quality scores (sequence only, unlike FASTQ)
- Represents the consensus genome after assembly
- Indexed with
.faifiles for fast random access by position
Genome Assembly
There are two classes of genome assembly from NGS data:
De Novo Assembly
- Latin for “from the new” — no reference sequence is used
- Results in contigs (contiguous sequences)
- Requires a scaffold to order contigs across repetitive regions
- Mostly used for metagenomics and assembling genomes with no available reference
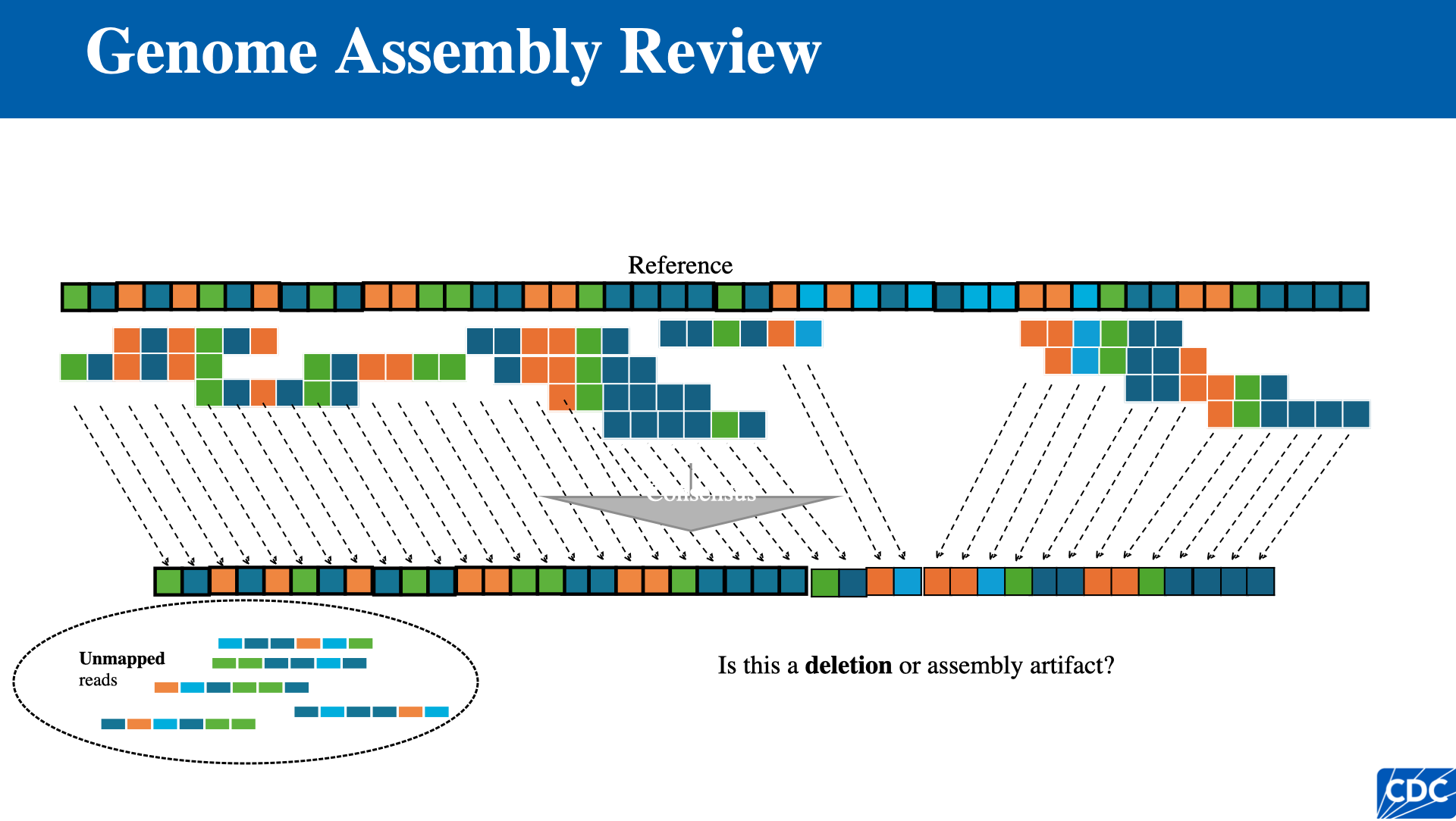
Reference-Based Assembly
- Individual reads are mapped directly to the position on the reference genome where they align best
- Disadvantage: highly variable or rapidly evolving pathogens may not assemble well if a close enough reference is unavailable
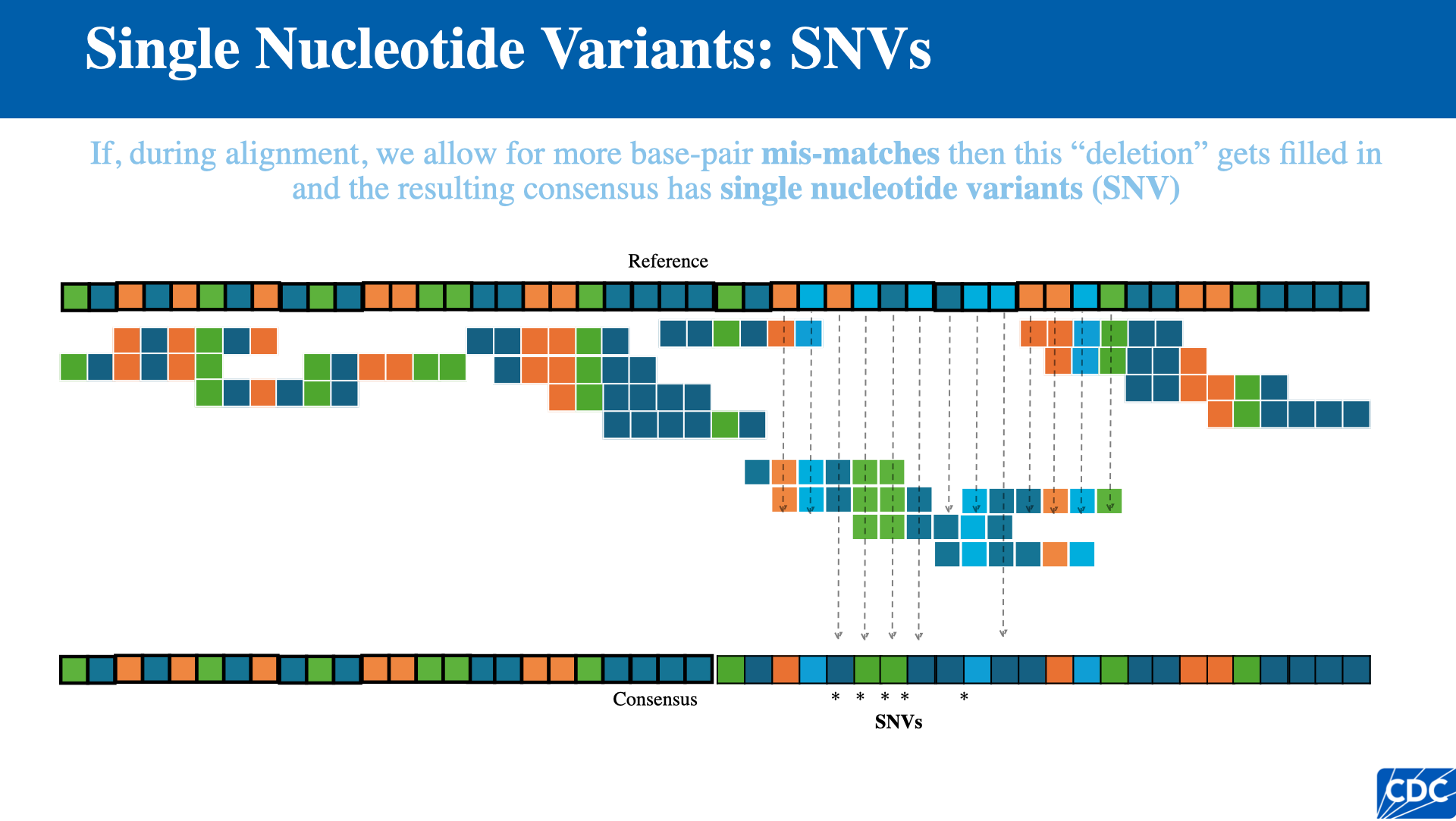
Single Nucleotide Variants (SNVs)
When aligning reads to a reference with tolerance for base-pair mismatches, positions where the consensus differs from the reference are called single nucleotide variants (SNVs).
IRMA: Iterative Refinement Meta-Assembler
Respiratory viruses are very diverse, and reference-based assembly works best when the right reference is chosen. Missing data (e.g., amplicon dropout) cannot be “reference-filled.”
The US CDC and all other WHO Collaborating Centres use IRMA for influenza genome assembly.
How IRMA Works
IRMA overcomes the problem of choosing an accurate reference sequence for assembling diverse influenza genomes through iterative refinement:
- Many type/subtype references are provided
- Each read is sorted to a reference based on top BLAT score
- A plurality consensus is built with SAM
- The new consensus is used as the reference to capture more reads with BLAT
- The read-gathering step is repeated (up to 5 times)
- Final assembly is repeated (up to 5 times)
- Plurality consensus is refined with SSW

From IRMA to MIRA
The IRMA algorithm is extensive, but a production pipeline also needs:
- Pre-assembly and post-assembly QC (trimming, downsampling, coverage, completeness, variant calls)
- Aggregation across runs
- Low-code options for analysts
- Additional features like genome annotation
MIRA (Molecular Influenza Resource Alignment) wraps IRMA with these capabilities.
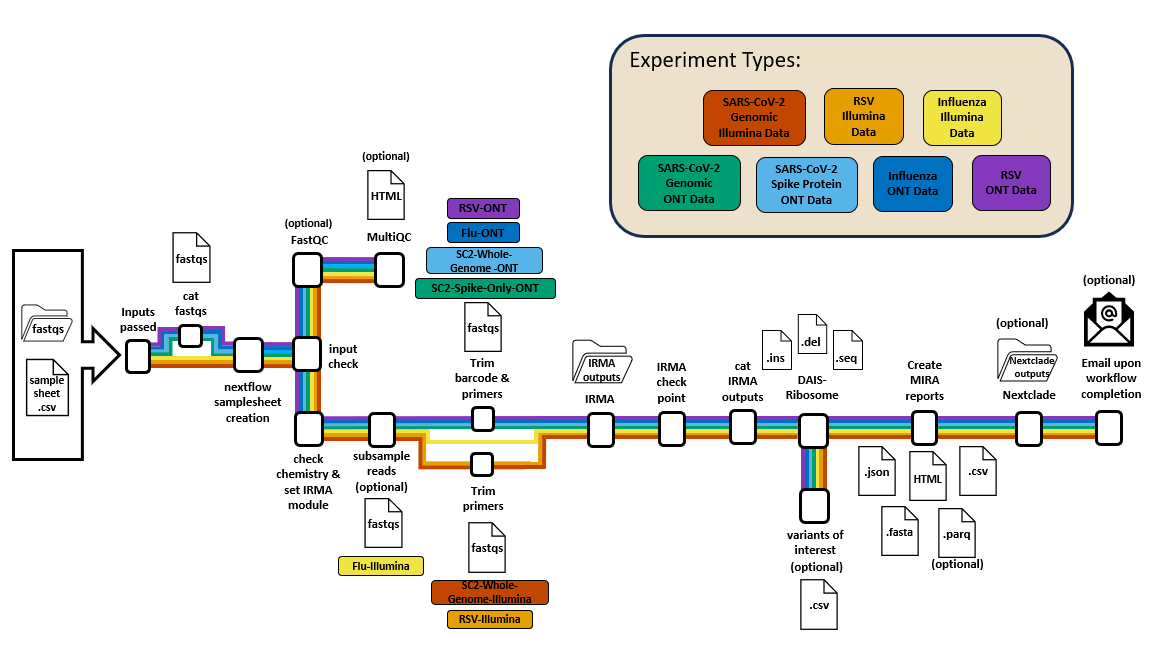
Workflow Managers
Workflow managers are essential for running complex bioinformatics pipelines:
- Automate complex pipelines — manage many steps, tools, and dependencies
- Track inputs, outputs, and dependencies — run only what needs updating
- Enable reproducibility — same workflow, same results across systems
- Scale from laptop to HPC/cloud — handle parallelization and scheduling
Popular Bioinformatics Workflow Managers
| Manager | Language | Notes |
|---|---|---|
| Snakemake (MIRA GUI) | Python-based, rule-driven | Familiar to Bash + Python users |
| Nextflow (MIRA-NF) | DSL built on Groovy | Strong HPC/scheduler integration |
Both integrate with containers via Docker/Singularity.
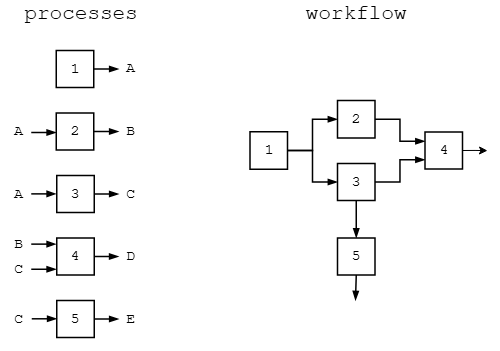
Figure credit: https://marcsingleton.github.io/posts/workflow-managers-in-data-science-nextflow-and-snakemake/
MIRA-NF
System Requirements and Setup
- Java v17 or higher (required by Nextflow)
- Nextflow workflow engine
- Singularity-CE or Docker for containerized execution
- Git for cloning the repository
-profile test,docker or -profile test,singularity) before running real data.Installation
Java Installation (WSL/Linux)
# Update packages
sudo apt update && sudo apt upgrade -y
# Install Java
sudo apt install openjdk-17-jdk -y
# Verify
java -version
Java Installation (macOS)
# Check if Java is installed
java -version
# Install Homebrew (if needed)
/bin/bash -c "$(curl -fsSL https://raw.githubusercontent.com/Homebrew/install/HEAD/install.sh)"
# Install Java
brew install openjdk
# Add to path
sudo ln -sfn /opt/homebrew/opt/openjdk/libexec/openjdk.jdk /Library/Java/JavaVirtualMachines/openjdk.jdk
Nextflow Installation
# Download and install
curl -s https://get.nextflow.io | bash
# Move to system path
sudo mv nextflow /usr/local/bin/
# Ensure executable
sudo chmod +x /usr/local/bin/nextflow
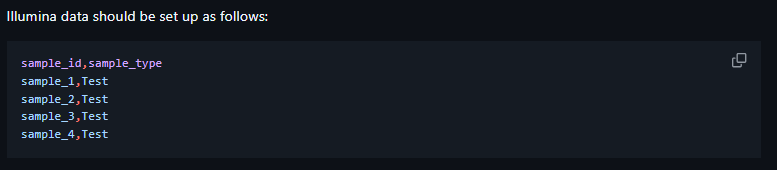
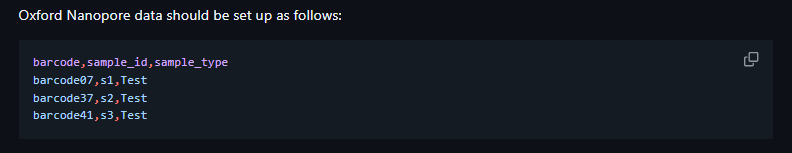
Input and Samplesheet Requirements
Prepare a samplesheet CSV with the following columns:
| Platform | Required columns |
|---|---|
| Illumina | sample_id, sample_type |
| ONT | barcode, sample_id, sample_type |
Directory Layout
FASTQ files and the samplesheet go in a run folder used as --runpath. The name of the run folder will be used to name output files.
sample_id column must be unique
- Headers must be named exactly as shown above
- No empty lines at the end of the samplesheet
- Illumina samples must have both R1 and R2 for all samples
- Illumina FASTQ naming: {sample_id}_R1*fastq* / {sample_id}_R1*fq* and corresponding R2
Running the Pipeline
Core Command
nextflow run ./main.nf \
-profile singularity,local \
--input <RUN_PATH>/samplesheet.csv \
--outdir <OUTDIR> \
--runpath <RUN_PATH> \
--e <EXPERIMENT_TYPE> \
[other optional flags]
| Parameter | Description |
|---|---|
-profile |
Compute environment (e.g., singularity, docker, local, slurm, sge) |
--e |
Experiment type (e.g., Flu-ONT, SC2-Whole-Genome-Illumina) |
Useful Optional Flags
| Flag | Description |
|---|---|
--p |
Built-in primer schema (for SC2/RSV) |
--custom_primers |
Supply custom primer FASTA |
--subsample_reads |
Limit reads for faster analysis |
--parquet_files |
Generate additional Parquet files (formatted like CSV) |
--read_qc |
Run FastQC/MultiQC modules |
--nextclade |
Run Nextclade on passing samples |
--process_q / --email |
HPC queue and notification options |
MIRA-NF Output Structure
outputs/
├── aggregate_outputs/
│ ├── multiqc/ # MultiQC outputs (when applicable)
│ ├── dais-ribosome/ # DAIS inputs and outputs
│ ├── dash-json/ # JSON files
│ ├── mira-reports/ # Aggregated FASTA and HTML files
│ ├── csv-reports/ # CSV summary files
│ └── parquet-reports/ # Parquet files (when applicable)
├── <Sample_ID>/
│ ├── subsampled-reads/ # FASTQs and log files (when applicable)
│ ├── barcode-trimmed-reads/# FASTQs and log files (when applicable)
│ ├── primer-trimmed-reads/ # FASTQs and log files (when applicable)
│ ├── IRMA/<Sample_ID>/ # IRMA outputs and log files
│ └── IRMA-negative/ # Negative results (when applicable)
├── nextclade/
│ ├── input_fasta_files/ # Input FASTAs for Nextclade
│ └── ... # Aligned FASTAs, Auspice JSON, CSV files
├── fastq_pass/ # ONT only — concatenated FASTQs
└── pipeline_info/ # Execution reports, sad_samples.tsv, versions
Each sample also contains all IRMA outputs per sample.
SAM and BAM Files
SAM Files (Sequence Alignment/Map)
SAM is a text-based alignment format that stores how reads align to a reference genome.
- Human-readable — can be viewed and inspected directly
- Contains alignments + metadata — read names, positions, flags, CIGAR strings, mapping quality
- Large file size — not efficient for storage or sharing
SAM Field Definitions
| Col | Field | Type | Description |
|---|---|---|---|
| 1 | QNAME | String | Query template name |
| 2 | FLAG | Int | Bitwise flag |
| 3 | RNAME | String | Reference sequence name |
| 4 | POS | Int | 1-based leftmost mapping position |
| 5 | MAPQ | Int | Mapping quality |
| 6 | CIGAR | String | CIGAR string |
| 7 | RNEXT | String | Reference name of the mate/next read |
| 8 | PNEXT | Int | Position of the mate/next read |
| 9 | TLEN | Int | Observed template length |
| 10 | SEQ | String | Segment sequence |
| 11 | QUAL | String | ASCII of Phred-scaled base quality + 33 |

CIGAR Strings
CIGAR strings describe how a read aligns to the reference, encoding matches, mismatches, insertions, and deletions.
- Stored in the SAM/BAM alignment record — one CIGAR string per aligned read
- Format: number + operation — indicates length and type of alignment operation
Common CIGAR Operations
| Operation | Meaning |
|---|---|
| M | Alignment match/mismatch |
| I | Insertion relative to reference |
| D | Deletion relative to reference |
| S | Soft clipping (read bases not aligned) |
| H | Hard clipping (bases removed) |
| N | Skipped region (e.g., introns) |
Example: 10M2I5M1D20M
| Component | Meaning |
|---|---|
10M |
10 aligned bases |
2I |
2 inserted bases |
5M |
5 aligned bases |
1D |
1 deleted base |
20M |
20 aligned bases |
BAM Files (Binary Alignment/Map)
BAM is the binary (compressed) version of SAM — same information, smaller size.
- Efficient for storage and analysis — faster to read and write than SAM
- Requires sorting and indexing —
.baiindex enables random access by genomic position and is necessary for viewing - Standard input for downstream tools — variant calling, visualization, QC
samtools
samtools is the standard command-line toolkit for SAM/BAM files, used across NGS workflows.
Key Operations
| Command | Purpose |
|---|---|
samtools view |
View and convert SAM ↔ BAM (-b flag for BAM output) |
samtools sort |
Sort alignments — required for many downstream tools |
samtools index |
Create .bai index — enables random access |
samtools flagstat |
Alignment summary statistics |
samtools stats |
Detailed metrics |
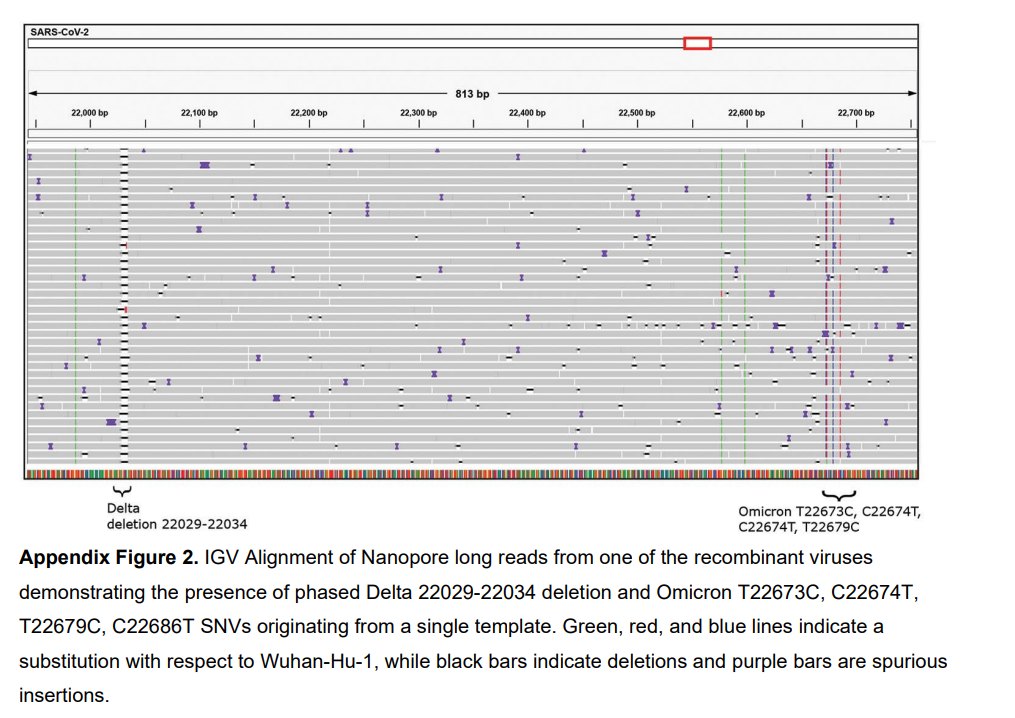
IGV: Integrative Genomics Viewer
IGV is an interactive tool for visualizing read alignments against a reference genome.
Requirements
- Sorted and indexed BAM files —
.bam+ corresponding.baiindex
Key Features
- Visualize read alignments — see how sequencing reads map across the genome
- Inspect alignment features — coverage depth, mismatches, indels, soft clipping, alignment gaps
- Navigate interactively — zoom from whole genome to single-base resolution; jump to coordinates or gene names

Common QC and Analysis Tasks in IGV
- Check alignment quality — consistent coverage and correct orientation
- Validate variants — confirm SNPs and indels are supported by reads
- Identify potential issues — low coverage regions, misalignments, primer artifacts, DI (defective interfering) particles
Multiple Sequence Alignment (MSA)
Multiple sequence alignment compares three or more sequences at once to identify conserved regions, variants, and evolutionary relationships. In influenza genomics, MSA is commonly used for:
- Comparing assembled HA or NA segments across samples
- Identifying gaps or insertions in assembled sequences
- Preparing input for phylogenetic analysis
A common command-line tool for MSA is MAFFT:
mafft input.fasta > aligned.fasta
Automating with Pipeline Scripts
MIRA-NF can be integrated into larger automation pipelines:
- Automate samplesheet creation
- Pipeline base-calling outputs into MIRA-NF
- Pipeline MIRA output tables to databases
- Pipeline output FASTAs to phylogenetic analyses
Next: Genome Assembly Practical for hands-on exercises using MIRA-NF.