Why Computer Architecture Matters for Bioinformatics
Content developed by Ben Rambo-Martin and Kristine Lacek
Bioinformatics workflows often involve:
- Processing gigabytes to terabytes of sequencing data
- Running computationally intensive algorithms)
- Managing memory for large reference genomes and indices
- Parallelizing tasks across multiple CPU cores
Understanding the underlying hardware helps you:
| Scenario | Knowledge Applied |
|---|---|
| Pipeline runs slowly | Is it CPU-bound, Memory “swapping” or I/O-bound? |
| “Out of memory” errors | How much RAM does the tool need? Can you use disk-based alternatives? |
| Choosing cloud instances | How many cores? How much memory? SSD vs HDD? |
| Optimizing tool parameters | CPU/Thread count, memory limits, temp directory location |
Module Objectives
- Basic computer architecture
- BIOS/UEFI
- Introduce Operating Systems (OS)
- Understand how to use Linux on Windows
- What is a virtual machine is?
- How do operating systems differ?
- What does *nix means for bioinformatics?
- Install WSL on windows machines, access command prompt from MacOS

Computer Architecture Overview
The diagram below shows the main components of a computer and how they interact:
The CPU (Central Processing Unit)
3.1 What the CPU does
The CPU executes instructions — the fundamental operations that make up all software. For bioinformatics, this includes:
- Comparing nucleotide sequences character by character
- Calculating alignment scores
- Evaluating quality score thresholds
3.2 Cores and threads
Modern CPUs have multiple cores — independent processing units that can work in parallel:
| Term | Definition | Bioinformatics relevance |
|---|---|---|
| Core | A physical processing unit | Each core can run one task at a time |
| Thread | A virtual execution stream | Hyperthreading gives 2 threads per core |
| Multi-threading | Using multiple threads | -t 8 in BWA-MEM uses 8 threads |
-t 4 for CPU-intensive tasks.
3.3 CPU cache hierarchy
Data must travel from RAM to the CPU to be processed. CPU caches are small, ultra-fast memory buffers that store frequently accessed data:
| Cache | Size (typical) | Speed | Purpose |
|---|---|---|---|
| L1 | 32-64 KB per core | Fastest | Currently executing instructions |
| L2 | 256-512 KB per core | Very fast | Recent data per core |
| L3 | 8-64 MB shared | Fast | Shared across all cores |
| RAM | 8-256+ GB | Slower | Main working memory |
The CPU automatically manages this hierarchy — when it needs data not in cache, it fetches it from RAM (a “cache miss”), which is slower.
3.4 Checking your CPU
Linux/WSL:
# Number of cores
nproc
# Detailed CPU info
lscpu
# Or from /proc
cat /proc/cpuinfo | grep "model name" | head -1
cat /proc/cpuinfo | grep "cpu cores" | head -1
macOS:
sysctl -n hw.ncpu # Total threads
sysctl -n hw.physicalcpu # Physical cores
Memory (RAM)
4.1 What RAM does
RAM holds data that the CPU is actively working with:
- The genome sequence being aligned against
- Index structures (FM-index, hash tables, dataframes)
- Reads currently being processed
When you load a reference genome into a tool like BWA, it gets loaded into RAM. The larger the reference or index, the more RAM required.
4.2 Common memory requirements
| Task | Typical RAM needed |
|---|---|
| Mira-influenza | 8-16 GB |
| Mira-sc2, Aligning to human genome (BWA-MEM2) | 16-32 GB |
| BEAST phylogenetics, De novo assembly (SPAdes, large genome) | 64-256+ GB |
| Metagenomics classification (Kraken2) | 8-64 GB (depends on database) |
4.3 Virtual memory and swap
When RAM runs out, the operating system uses virtual memory — disk space that pretends to be RAM:
- Also called “swap” (Linux) or “page file” (Windows)
- Much slower than real RAM (100-1000x slower)
- Causes severe performance degradation (“thrashing”)
4.4 Checking memory usage
Linux/WSL:
# Quick overview
free -h
# Detailed memory info
cat /proc/meminfo | head -10
# Watch memory in real-time
htop # or: watch -n 1 free -h
macOS:
# Memory summary
vm_stat
# More readable
top -l 1 | head -n 10
4.5 Reducing memory usage
When RAM is limited:
- Use streaming algorithms — Process reads one at a time instead of loading all into memory
- Reduce thread count — Each thread may need its own memory buffer
- Use disk-based alternatives — Some tools offer memory-efficient modes
- Split input files — Process in chunks, merge results
- Use a cluster/cloud — Rent machines with more RAM
Storage (Disk)
5.1 Storage types
| Type | Speed | Cost |
|---|---|---|
| NVMe SSD | 3,000-7,000 MB/s | $$$ |
| SATA SSD | 500-600 MB/s | $$ |
| HDD | 100-200 MB/s | $ |
| Network storage | Variable (1-1000 MB/s) | Variable |
5.2 Impact on bioinformatics
Many workflows are I/O bound — they spend more time reading/writing data than computing:
- Writing millions of SAM/BAM alignment records
- Reading large FASTQ files
- Sorting and indexing BAM files
- Building and querying databases
5.3 Checking disk space and speed
Linux/WSL:
# Disk space
df -h
# Which disk a directory is on
df -h /path/to/directory
# Simple write speed test
dd if=/dev/zero of=testfile bs=1G count=1 oflag=direct 2>&1 | tail -1
# Clean up
rm testfile
5.4 Storage best practices
- Use SSDs for temp files — Set
$TMPDIRor tool-specific temp directories to SSD - Compress when possible — Gzipped FASTQ takes 3-4x less space and often process faster
- Clean up intermediate files — BAM files from failed runs, temp files, etc.
- Archive completed projects — Move to cheaper HDD or tape storage
Operating System

6.1 Role of the OS
The operating system:
- Manages memory — Allocates RAM to programs, handles virtual memory
- Schedules CPU — Decides which processes run on which cores
- Handles I/O — Coordinates disk reads/writes, network traffic
- Provides interfaces — File systems, command line, software installation
6.2 Linux dominance in bioinformatics
Most bioinformatics tools are designed for Linux:
| OS | Bioinformatics support |
|---|---|
| Linux (Ubuntu, CentOS, etc.) | Excellent — most tools native |
| macOS | Good — Unix-based, most tools work |
| Windows | Limited — use WSL for Linux tools |
6.3 Key OS concepts
Command Line Interface:
Much of bioinformatics is done in a Linux (or Unix) OS — collectively called *nix. Unix and Linux are built around a command-line–focused design for system control, automation, and scripting.
CLI Terminology:
- Shell — The program that interprets command-line input and runs commands (bash, zsh, PowerShell)
- Prompt — Text displayed by the shell indicating it is ready to accept a command
- Command — A program or instruction typed into the shell to perform an action
- Flag (or option) — A modifier added to a command that changes how it behaves
- Directory — A location in the filesystem used to organize files
- Path — The exact location of a file or directory in the filesystem
- Permissions — Rules that control who can read, write, or execute a file or directory
Linux Filesystem:
Linux treats most things like files — one consistent way to access resources:
- Regular files (documents, programs)
- Directories
- Devices (
/dev) - System information (
/proc)
All files exist under one root directory:
/
├── home/
├── etc/
├── var/
└── dev/
File systems:
- Linux uses paths like
/home/user/data/ - Windows uses paths like
C:\Users\user\data\ - WSL bridges both:
/mnt/c/Users/accesses Windows files from Linux
Environment variables:
$PATH— Where the shell looks for executables$HOME— Your home directory$TMPDIR— Where temporary files go
Package managers:
- conda/mamba — Cross-platform, recommended for bioinformatics
- apt (Ubuntu/Debian) — System packages
- brew (macOS) — macOS packages
6.4 Checking system info
# OS version
cat /etc/os-release # Linux
sw_vers # macOS
# Kernel version
uname -r
# All system info summary
neofetch # if installed, fun visualization
Putting It All Together
Bioinformatics and the Command Line

7.1 Minimum specs for bioinformatics
| Component | Minimum | Recommended | Heavy workloads |
|---|---|---|---|
| CPU | 4 cores | 8+ cores | 16-32+ cores |
| RAM | 8 GB | 32 GB | 64-256+ GB |
| Storage | 256 GB SSD | 1 TB SSD | 2+ TB NVMe SSD |
7.2 Diagnosing performance issues
When something runs slowly, ask:
- Is CPU at 100%? → CPU-bound, consider more cores or faster algorithm
- Is RAM full / swapping? → Memory-bound, reduce usage or get more RAM
- Is disk activity constant but CPU idle? → I/O-bound, use faster storage
- Is network activity the bottleneck? → Download data locally first
Monitoring tools:
htop # Interactive process viewer (CPU, RAM per process)
iotop # Disk I/O by process (requires sudo)
nmon # All-in-one system monitor
7.3 When to use cloud/cluster
Consider external compute when:
- Job needs more RAM than your machine has
- Job would take days on your laptop
- You need to run many jobs in parallel
- You need specialized hardware (GPUs for ML)
Common platforms:
- Institutional HPC clusters — Often “free” for researchers
- AWS, Google Cloud, Azure — Pay per hour, flexible
7.4 Summary checklist
Before running a pipeline:
- Do I have enough RAM for the largest step?
- Do I have enough disk space for inputs, outputs, and temp files?
- Is my working directory on fast storage (SSD)?
- Have I set appropriate thread counts for my CPU?
- Do I know where temp files will be written?
Networks and Servers
A network is a system that connects computers and devices, allowing them to share data, resources, and services through wired or wireless communication links.
A server is a computer system designed to provide resources or services—such as data, applications, or network functions—to other devices over a network.
- Optimized for reliability and continuous operation
- Handle requests from clients, managing tasks:
- Data storage
- Application hosting
- Authentication
- Communication between systems
Practical: Command-Line on Your Computer
macOS
- Press Command (⌘) + Spacebar to open Spotlight
- Type Terminal
- Press Return to open it
- A command prompt will appear on your screen
Windows
- Run PowerShell as Administrator
- Run the following command in PowerShell:
wsl --install - Restart your computer
- Reopen PowerShell and enter the following commands:
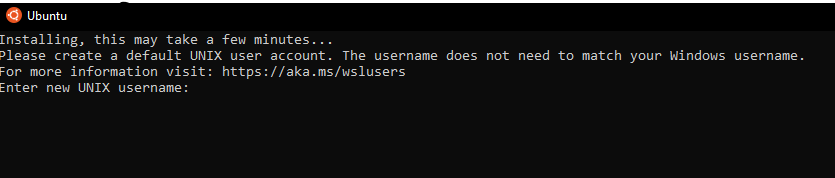
wsl --set-default-version 2 wsl --install -d Ubuntu-24.04 - Following successful installation, an Ubuntu terminal should pop up
- Enter a username that will be exclusive for WSL. Press Enter and then enter a password
- A command prompt will appear on your screen
Linux
- Open the Applications or Activities menu (top-left or bottom-left, depending on your system)
- Search for Terminal, Console, or Xterm (names vary by distribution)
- Click the icon to launch it
- A command prompt will appear on your screen