Hierarchical Latent Infections: Understanding Temporal Process Choices
Code
import jax.numpy as jnp
import jax.random as random
import numpy as np
import numpyro
from numpyro.infer import Predictive
import pandas as pd
import plotnine as p9
import warnings
warnings.filterwarnings("ignore")
from _tutorial_theme import theme_tutorial
Code
from pyrenew.latent import (
HierarchicalInfections,
AR1,
DifferencedAR1,
RandomWalk,
)
from pyrenew.deterministic import DeterministicPMF, DeterministicVariable
The Modeling Problem
You have a jurisdiction (e.g., a state or metropolitan area) with multiple subpopulations. Some subpopulations have direct observations; others do not. Disaggregated data from many subpopulations is often both sparse and noisy. You want to model transmission dynamics that:
- Share information across subpopulations (pooling strength)
- Allow local transmission to deviate from the jurisdiction average
- Produce estimates for both observed and unobserved areas
HierarchicalInfections addresses this by decomposing the effective
reproduction number \(\mathcal{R}(t)\) into:
where:
- \(\mathcal{R}_{\text{baseline}}(t)\) is the jurisdiction-level reproduction number (shared across all subpopulations)
- \(\delta_k(t)\) is the deviation for subpopulation \(k\), constrained so that \(\sum_k \delta_k(t) = 0\)
The sum-to-zero constraint ensures \(\mathcal{R}_{\text{baseline}}(t)\) represents the geometric mean of subpopulation \(\mathcal{R}\) values—a natural “center” for the hierarchy.
Each subpopulation runs its own renewal equation with its own \(\mathcal{R}_k(t)\), then results are aggregated for jurisdiction-level quantities (like total hospital admissions).
Model Inputs
HierarchicalInfections requires several inputs that shape the
infection dynamics. Understanding what each does helps you configure the
model appropriately.
A Note on RandomVariables
All inputs to HierarchicalInfections are RandomVariables—objects
that can be sampled. In this tutorial, we use DeterministicVariable
and DeterministicPMF (fixed values) for clarity. In real inference,
you would typically use DistributionalVariable with priors for
quantities you want to estimate (like I0 or initial Rt).
For example:
# Fixed value (for illustration)
I0_rv = DeterministicVariable("I0", 0.001)
# With a prior (for inference)
from pyrenew.randomvariable import DistributionalVariable
import numpyro.distributions as dist
I0_rv = DistributionalVariable("I0", dist.Beta(1, 1000))
Population Structure
We define subpopulations as fractions of the total jurisdiction population:
Code
# 6 subpopulations with their population fractions
# Which subpopulations are "observed" or "unobserved" is determined by each
# observation process (via subpop_indices), not by the latent process.
subpop_fractions = jnp.array([0.10, 0.14, 0.21, 0.22, 0.07, 0.26])
n_subpops = len(subpop_fractions)
print(f"Number of subpopulations: {n_subpops}")
print(f"Total population fraction: {float(jnp.sum(subpop_fractions)):.2f}")
Number of subpopulations: 6
Total population fraction: 1.00
Note: Population structure is passed at sample time, not when constructing the process. This allows the same model specification to be fit to different jurisdictions.
Generation Interval
The generation interval is the probability distribution over the time between infection of a primary case and infection of a secondary case. It determines the “memory” of the renewal process—how current infections depend on past infections.
Code
# Typical COVID-like generation interval
gen_int_pmf = jnp.array([0.16, 0.32, 0.25, 0.14, 0.07, 0.04, 0.02])
gen_int_rv = DeterministicPMF("gen_int", gen_int_pmf)
days = np.arange(len(gen_int_pmf))
mean_gen_time = float(np.sum(days * gen_int_pmf))
print(f"Mean generation time: {mean_gen_time:.1f} days")
Mean generation time: 1.8 days
What it controls: The generation interval affects how quickly infection dynamics respond to changes in Rt:
- Shorter generation interval → faster response, more volatile dynamics
- Longer generation interval → slower response, smoother dynamics
The generation interval is typically fixed based on pathogen biology, not estimated from data.
Code
# Compare generation intervals: short vs long
gi_short = jnp.array([0.4, 0.4, 0.2]) # Mean ~1 day
gi_long = jnp.array([0.05, 0.1, 0.2, 0.3, 0.2, 0.1, 0.05]) # Mean ~3.5 days
gi_short_rv = DeterministicPMF("gi_short", gi_short)
gi_long_rv = DeterministicPMF("gi_long", gi_long)
print(
f"Short GI mean: {float(np.sum(np.arange(len(gi_short)) * gi_short)):.1f} days"
)
print(
f"Long GI mean: {float(np.sum(np.arange(len(gi_long)) * gi_long)):.1f} days"
)
Short GI mean: 0.8 days
Long GI mean: 3.0 days
To illustrate, we sample infection trajectories using each generation
interval while holding other inputs fixed (I0 = 0.001, DifferencedAR1
baseline, AR1 deviations) and using the same random seed so the
underlying Rt trajectory is identical. Solid lines use
initial_log_rt = 0 (Rt starts at 1); dashed lines show
initial_log_rt = ±0.2 (Rt starts growing or declining) with the
standard GI:
Code
# Create models with different generation intervals
baseline_rt_process = DifferencedAR1(autoreg=0.9, innovation_sd=0.05)
subpop_rt_deviation_process = AR1(autoreg=0.8, innovation_sd=0.05)
n_days_demo = 28
gi_data = []
# GI comparison at initial_log_rt = 0
gi_configs = {
"Short GI (~1 day)": gi_short_rv,
"Standard GI (~2 days)": gen_int_rv,
"Long GI (~3.5 days)": gi_long_rv,
}
# Use consistent n_initialization_points across all GI configs
# so the temporal process trajectories are comparable (same seed → same Rt)
n_init_gi = max(len(rv()) for rv in gi_configs.values())
for name, gi_rv in gi_configs.items():
model = HierarchicalInfections(
gen_int_rv=gi_rv,
I0_rv=DeterministicVariable("I0", 0.001),
initial_log_rt_rv=DeterministicVariable("initial_log_rt", 0.0),
baseline_rt_process=baseline_rt_process,
subpop_rt_deviation_process=subpop_rt_deviation_process,
n_initialization_points=n_init_gi,
)
with numpyro.handlers.seed(rng_seed=42):
inf_agg, _ = model.sample(
n_days_post_init=n_days_demo,
subpop_fractions=subpop_fractions,
)
n_init = model.n_initialization_points
inf = np.array(inf_agg)[n_init:]
for d in range(len(inf)):
gi_data.append(
{
"day": d,
"infections": float(inf[d]),
"config": name,
"linetype": "solid",
}
)
# initial_log_rt comparison with standard GI
for init_rt_name, init_rt_val in [
("Rt≈1.22 (growing)", 0.2),
("Rt≈0.82 (declining)", -0.2),
]:
model = HierarchicalInfections(
gen_int_rv=gen_int_rv,
I0_rv=DeterministicVariable("I0", 0.001),
initial_log_rt_rv=DeterministicVariable("initial_log_rt", init_rt_val),
baseline_rt_process=baseline_rt_process,
subpop_rt_deviation_process=subpop_rt_deviation_process,
n_initialization_points=n_init_gi,
)
with numpyro.handlers.seed(rng_seed=42):
inf_agg, _ = model.sample(
n_days_post_init=n_days_demo,
subpop_fractions=subpop_fractions,
)
n_init = model.n_initialization_points
inf = np.array(inf_agg)[n_init:]
for d in range(len(inf)):
gi_data.append(
{
"day": d,
"infections": float(inf[d]),
"config": f"Standard GI, {init_rt_name}",
"linetype": "dashed",
}
)
gi_df = pd.DataFrame(gi_data)
(
p9.ggplot(
gi_df,
p9.aes(x="day", y="infections", color="config", linetype="config"),
)
+ p9.geom_line(size=1)
+ p9.scale_linetype_manual(
values={
"Short GI (~1 day)": "solid",
"Standard GI (~2 days)": "solid",
"Long GI (~3.5 days)": "solid",
"Standard GI, Rt≈1.22 (growing)": "dashed",
"Standard GI, Rt≈0.82 (declining)": "dashed",
}
)
+ p9.labs(
x="Days",
y="Infections (proportion)",
title="Effect of Generation Interval and Initial Rt",
color="Configuration",
linetype="Configuration",
)
+ theme_tutorial
+ p9.theme(legend_position="right")
)
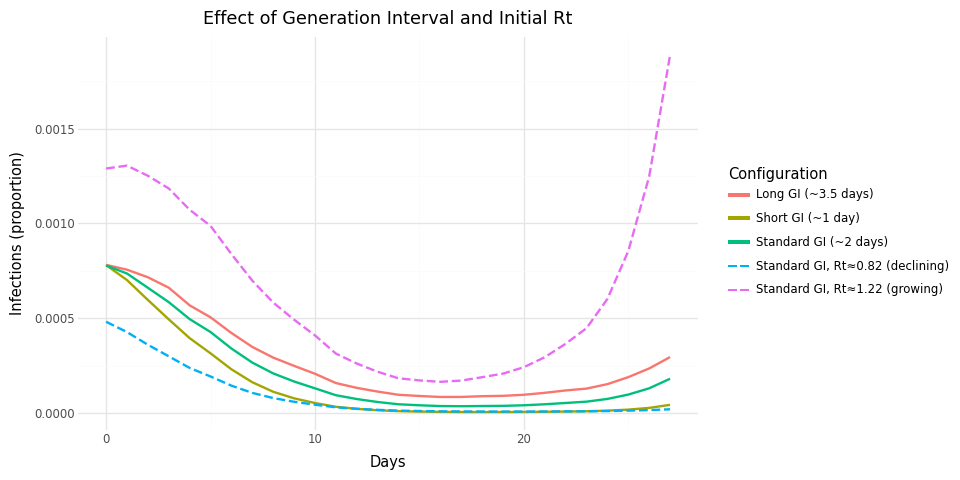
Figure 1: Effect of generation interval and initial_log_rt on infection dynamics. Solid lines start at Rt=1; dashed lines show initial growth (Rt≈1.22) or decline (Rt≈0.82).
Code
gi_df_zoom = gi_df[(gi_df["day"] >= 14) & (gi_df["day"] <= 21)]
(
p9.ggplot(
gi_df_zoom,
p9.aes(x="day", y="infections", color="config", linetype="config"),
)
+ p9.geom_line(size=1)
+ p9.scale_linetype_manual(
values={
"Short GI (~1 day)": "solid",
"Standard GI (~2 days)": "solid",
"Long GI (~3.5 days)": "solid",
"Standard GI, Rt≈1.22 (growing)": "dashed",
"Standard GI, Rt≈0.82 (declining)": "dashed",
}
)
+ p9.labs(
x="Days",
y="Infections (proportion)",
title="Zoomed: Days 14-21",
color="Configuration",
linetype="Configuration",
)
+ theme_tutorial
+ p9.theme(legend_position="right")
)
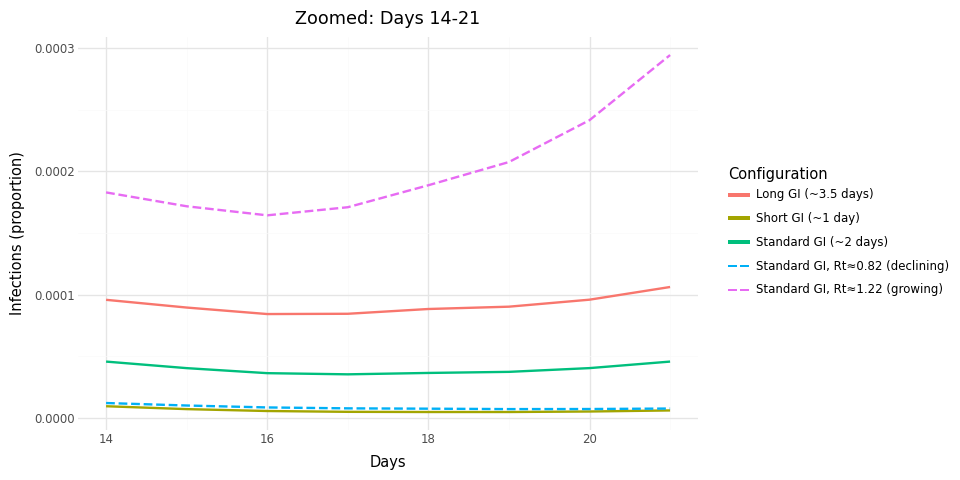
Figure 2: Zoomed view of days 14-21 showing separation between trajectories.
Initial Conditions: I0 and initial_log_rt
Parameters I0 and initial_log_rt set the starting point for the
infection process.
The previous plot shows the effect of initial_log_rt: the solid lines
use initial_log_rt = 0 (Rt starts at 1), while the dashed lines use
initial_log_rt = ±0.2 (Rt starts above or below 1). Notice that the
solid lines stay flat for the first ~15 days — when Rt = 1, each
infected person infects approximately one other, so infection levels
remain stable. The dashed lines show immediate growth or decline because
Rt starts away from 1.
The flat period for solid lines also reflects DifferencedAR1 with high
autoregression (0.9): this process models the rate of change of log(Rt),
starting from zero, and changes accumulate gradually. If you need more
initial dynamics, set initial_log_rt ≠ 0, use RandomWalk instead, or
increase innovation_sd.
I0 is the proportion of the population infected at time zero. It seeds
the renewal equation and scales the entire trajectory:
Code
# Default values
I0_rv = DeterministicVariable("I0", 0.001) # 0.1% initially infected
initial_log_rt_rv = DeterministicVariable(
"initial_log_rt", 0.0
) # Rt starts at 1.0
print(f"I0 = 0.001 means {0.001:.1%} of population initially infected")
print(f"initial_log_rt = 0.0 means Rt starts at {np.exp(0.0):.1f}")
I0 = 0.001 means 0.1% of population initially infected
initial_log_rt = 0.0 means Rt starts at 1.0
In the previous plot, I0 was specified as:
I0_rv=DeterministicVariable("I0", 0.001), i.e., \(0.1%\) of the
population. We overplot trajectories for \(10%\) and \(0.01%\) as well.
Code
i0_configs = {
"I0 = 0.0001 (0.01%)": 0.0001,
"I0 = 0.001 (0.1%)": 0.001,
"I0 = 0.01 (1%)": 0.01,
}
i0_results = {}
for name, i0_val in i0_configs.items():
model = HierarchicalInfections(
gen_int_rv=gen_int_rv,
I0_rv=DeterministicVariable("I0", i0_val),
initial_log_rt_rv=DeterministicVariable("initial_log_rt", 0.0),
baseline_rt_process=baseline_rt_process,
subpop_rt_deviation_process=subpop_rt_deviation_process,
n_initialization_points=len(gen_int_pmf),
)
with numpyro.handlers.seed(rng_seed=42):
inf_agg, _ = model.sample(
n_days_post_init=n_days_demo,
subpop_fractions=subpop_fractions,
)
n_init = model.n_initialization_points
i0_results[name] = np.array(inf_agg)[n_init:]
i0_data = []
for name, inf in i0_results.items():
for d in range(len(inf)):
i0_data.append({"day": d, "infections": float(inf[d]), "I0": name})
i0_df = pd.DataFrame(i0_data)
(
p9.ggplot(i0_df, p9.aes(x="day", y="infections", color="I0"))
+ p9.geom_line(size=1)
+ p9.scale_y_log10()
+ p9.labs(
x="Days",
y="Infections (proportion, log scale)",
title="Effect of Initial Infection Prevalence (I0)",
subtitle="Same Rt trajectory, different starting prevalence",
)
+ theme_tutorial
)
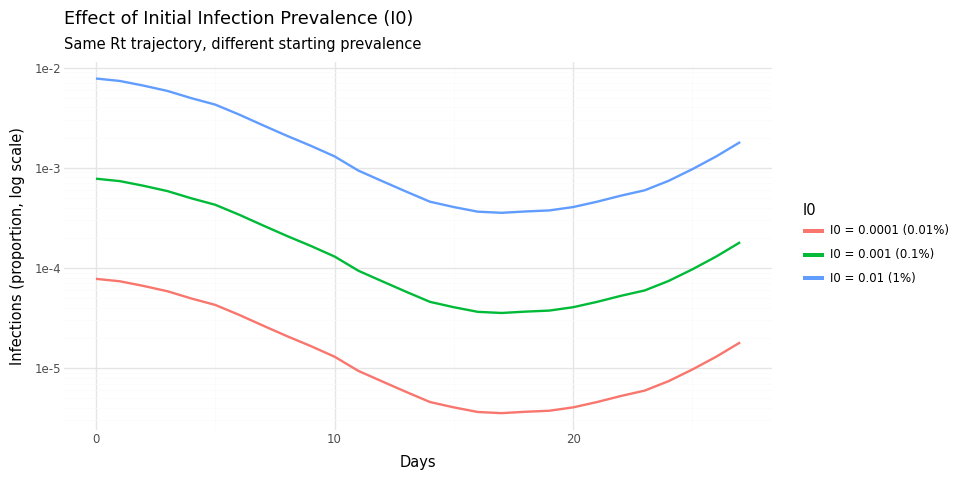
Figure 3: Effect of I0 on infection scale. Higher I0 shifts the entire trajectory up proportionally.
Temporal Processes
The temporal process determines how \(\mathcal{R}_{\text{baseline}}(t)\) and the deviations \(\delta_k(t)\) evolve over time. PyRenew provides three options:
| Process | What It Does | Epidemiological Interpretation |
|---|---|---|
RandomWalk |
Each value = previous + noise | Maximum flexibility; Rt can drift anywhere |
AR1 |
Each value reverts toward a mean | Rt stays bounded near a baseline; deviations decay |
DifferencedAR1 |
The rate of change reverts toward zero | Rt can trend persistently, but growth rate stabilizes |
Choosing a temporal process:
| If you believe… | Consider |
|---|---|
| Rt should stay near 1 (endemic equilibrium) | AR1 for baseline |
| Rt may trend up/down over your horizon | DifferencedAR1 for baseline |
| Local differences are transient | AR1 for deviations |
| Local differences persist | RandomWalk for deviations |
Parameters:
autoreg(AR1, DifferencedAR1): How strongly the process reverts. Values near 1 = slow reversion; near 0 = fast.innovation_sd: Daily volatility. Larger values = more variable trajectories.
Configuring the Latent Process
Now let’s assemble all the inputs:
Code
# Baseline Rt: DifferencedAR1 allows trends while stabilizing growth rate
baseline_rt_process = DifferencedAR1(autoreg=0.9, innovation_sd=0.05)
# Subpopulation deviations: AR1 shrinks differences back toward zero
subpop_rt_deviation_process = AR1(autoreg=0.8, innovation_sd=0.05)
Why these choices?
DifferencedAR1for baseline: We don’t want to force Rt back to 1; epidemics can grow or decline persistently. But we do want the rate of change to stabilize.AR1for deviations: We expect local differences to be somewhat transient. If one area has a temporary spike, it should eventually return toward the jurisdiction average.
Creating the Process
Code
latent_infections = HierarchicalInfections(
gen_int_rv=gen_int_rv,
I0_rv=I0_rv,
initial_log_rt_rv=initial_log_rt_rv,
baseline_rt_process=baseline_rt_process,
subpop_rt_deviation_process=subpop_rt_deviation_process,
n_initialization_points=len(gen_int_pmf),
)
print(
f"Generation interval length: {latent_infections.get_required_lookback()} days"
)
print(
f"Initialization points: {latent_infections.n_initialization_points} days"
)
Generation interval length: 7 days
Initialization points: 7 days
Prior Predictive Checks: What Does the Model Believe?
Now we examine what infection dynamics this configuration considers plausible—before seeing any data.
Single Sample
Code
n_days = 28 # 4 weeks of simulation
with numpyro.handlers.seed(rng_seed=42):
result = latent_infections.sample(
n_days_post_init=n_days,
subpop_fractions=subpop_fractions,
)
infections_agg, infections_all = result
n_total = infections_agg.shape[0]
n_init = n_total - n_days
print("Output shapes:")
print(f" Aggregate (jurisdiction total): {infections_agg.shape}")
print(f" All subpopulations: {infections_all.shape}")
print(
f"\nTimeline: {n_init} initialization days + {n_days} simulation days = {n_total} total"
)
Output shapes:
Aggregate (jurisdiction total): (35,)
All subpopulations: (35, 6)
Timeline: 7 initialization days + 28 simulation days = 35 total
Many Samples: The Prior Predictive Distribution
A single sample tells us little. We need many samples to understand the distribution of trajectories the model considers plausible.
Code
def sample_model():
"""Wrapper for prior predictive sampling."""
return latent_infections.sample(
n_days_post_init=n_days,
subpop_fractions=subpop_fractions,
)
n_samples = 200
predictive = Predictive(sample_model, num_samples=n_samples)
prior_samples = predictive(random.PRNGKey(42))
print(
f"Rt baseline samples: {prior_samples['latent_infections::rt_baseline'].shape}"
)
print(
f"Infection samples: {prior_samples['latent_infections::infections_aggregate'].shape}"
)
Rt baseline samples: (200, 35, 1)
Infection samples: (200, 35)
Visualizing the Prior: Baseline Rt Trajectories
Code
# Extract post-initialization Rt trajectories
rt_samples = np.array(prior_samples["latent_infections::rt_baseline"])[
:, n_init:, 0
]
# Build long-format dataframe for plotting
rt_data = []
for i in range(n_samples):
for d in range(n_days):
rt_data.append({"day": d, "rt": float(rt_samples[i, d]), "sample": i})
rt_df = pd.DataFrame(rt_data)
# Compute summary statistics
rt_median = np.median(rt_samples, axis=0)
rt_mean = np.mean(rt_samples, axis=0)
rt_q05 = np.percentile(rt_samples, 5, axis=0)
rt_q95 = np.percentile(rt_samples, 95, axis=0)
Code
# Cap display for readability (some trajectories explode)
rt_cap = 5.0
n_extreme = np.sum(np.any(rt_samples > rt_cap, axis=1))
(
p9.ggplot(rt_df, p9.aes(x="day", y="rt", group="sample"))
+ p9.geom_line(alpha=0.1, size=0.4, color="steelblue")
+ p9.geom_hline(yintercept=1.0, color="red", linetype="dashed", alpha=0.7)
+ p9.scale_x_continuous(expand=(0, 0))
+ p9.coord_cartesian(ylim=(0, rt_cap))
+ p9.labs(
x="Days",
y="Rt (baseline)",
title=f"Prior Predictive: Baseline Rt ({n_samples} samples)",
subtitle=f"Red dashed line: Rt = 1. {n_extreme} samples exceed Rt = {rt_cap} (not shown in full).",
)
+ theme_tutorial
)
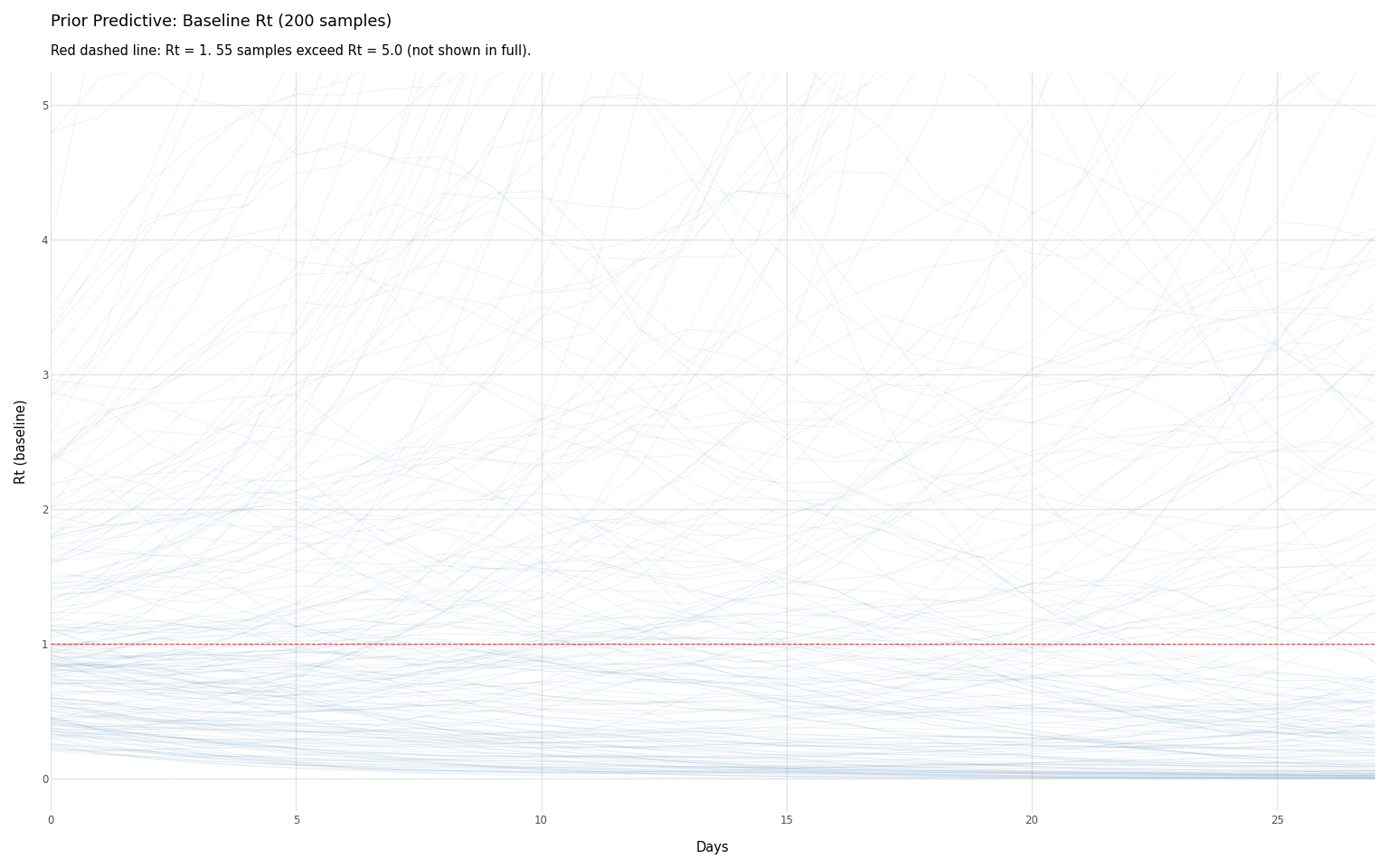
Figure 4: Prior predictive distribution of baseline Rt. Each light blue line is one sample from the prior. The model considers all these trajectories plausible before seeing data.
Interpreting the Prior Predictive
Code
print("Rt at end of simulation (day 28):")
print(f" 5th percentile: {rt_q05[-1]:.2f}")
print(f" Median: {rt_median[-1]:.2f}")
print(f" Mean: {rt_mean[-1]:.2f}")
print(f" 95th percentile: {rt_q95[-1]:.2f}")
print(f" Maximum: {np.max(rt_samples[:, -1]):.1f}")
Rt at end of simulation (day 28):
5th percentile: 0.01
Median: 0.82
Mean: 9.68
95th percentile: 41.50
Maximum: 364.0
What this tells us:
The median stays near 1, but the mean is pulled up by explosive trajectories. This is a consequence of modeling on the log scale:
- Half the trajectories have Rt \< 1 (bounded below by 0)
- Half have Rt > 1 (unbounded above—can reach 10, 50, 100+)
- The explosive trajectories dominate the mean
Is this a problem? It depends on your data. With abundant data, these extreme trajectories will be ruled out by the likelihood. With sparse data, they may not be—and your posterior could include implausible dynamics.
Comparing Temporal Process Choices
The prior predictive distribution changes dramatically based on your temporal process choices. Let’s compare the three options for baseline Rt.
Code
temporal_configs = {
"AR(1)": AR1(autoreg=0.9, innovation_sd=0.05),
"DifferencedAR1": DifferencedAR1(autoreg=0.5, innovation_sd=0.05),
"RandomWalk": RandomWalk(innovation_sd=0.05),
}
process_samples = {}
for name, temporal in temporal_configs.items():
model = HierarchicalInfections(
gen_int_rv=gen_int_rv,
I0_rv=I0_rv,
initial_log_rt_rv=initial_log_rt_rv,
baseline_rt_process=temporal,
subpop_rt_deviation_process=AR1(autoreg=0.8, innovation_sd=0.05),
n_initialization_points=len(gen_int_pmf),
)
def make_sampler(m):
return lambda: m.sample(
n_days_post_init=n_days,
subpop_fractions=subpop_fractions,
)
samples = Predictive(make_sampler(model), num_samples=n_samples)(
random.PRNGKey(42)
)
process_samples[name] = np.array(
samples["latent_infections::rt_baseline"]
)[:, n_init:, 0]
Code
# Build comparison dataframe
comparison_data = []
for name, rt in process_samples.items():
for i in range(n_samples):
for d in range(n_days):
comparison_data.append(
{
"day": d,
"rt": float(rt[i, d]),
"sample": i,
"process": name,
}
)
comparison_df = pd.DataFrame(comparison_data)
comparison_df["process"] = pd.Categorical(
comparison_df["process"],
categories=["AR(1)", "DifferencedAR1", "RandomWalk"],
ordered=True,
)
Code
(
p9.ggplot(comparison_df, p9.aes(x="day", y="rt", group="sample"))
+ p9.geom_line(alpha=0.1, size=0.4, color="steelblue")
+ p9.geom_hline(yintercept=1.0, color="red", linetype="dashed", alpha=0.7)
+ p9.facet_wrap("~ process", ncol=3)
+ p9.scale_x_continuous(expand=(0, 0))
+ p9.coord_cartesian(ylim=(0, 3.0))
+ p9.labs(
x="Days",
y="Rt (baseline)",
title="Prior Predictive by Temporal Process",
subtitle="Same innovation_sd=0.05; AR(1) autoreg=0.9, DifferencedAR1 autoreg=0.5",
)
+ theme_tutorial
)
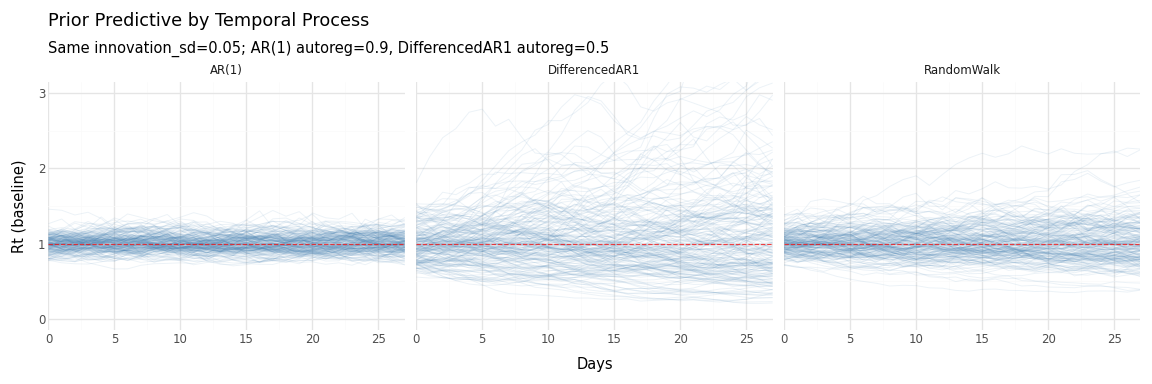
Figure 5: Effect of temporal process choice on prior Rt trajectories. AR(1) stays bounded near 1; DifferencedAR1 and RandomWalk can drift further.
Code
print("Rt at day 28 by temporal process:")
print(f"{'Process':<18} {'Median':>8} {'Mean':>8} {'95th %':>8} {'Max':>8}")
print("-" * 52)
for name in ["AR(1)", "DifferencedAR1", "RandomWalk"]:
rt = process_samples[name][:, -1]
print(
f"{name:<18} {np.median(rt):>8.2f} {np.mean(rt):>8.2f} "
f"{np.percentile(rt, 95):>8.2f} {np.max(rt):>8.1f}"
)
Rt at day 28 by temporal process:
Process Median Mean 95th % Max
----------------------------------------------------
AR(1) 1.01 1.01 1.23 1.3
DifferencedAR1 1.04 1.16 2.44 3.9
RandomWalk 0.99 1.02 1.51 2.3
Key observations:
- AR(1) produces the tightest distribution—Rt reverts toward 1
- DifferencedAR1 allows more drift but growth rate stabilizes
- RandomWalk has the widest spread—no mean reversion at all
Subpopulation Deviations
The same choice applies to subpopulation deviations \(\delta_k(t)\). Let’s compare AR(1) vs RandomWalk:
Code
subpop_configs = {
"AR(1) deviations": AR1(autoreg=0.8, innovation_sd=0.05),
"RandomWalk deviations": RandomWalk(innovation_sd=0.05),
}
deviation_samples = {}
for name, subpop_rt_deviation_process in subpop_configs.items():
model = HierarchicalInfections(
gen_int_rv=gen_int_rv,
I0_rv=I0_rv,
initial_log_rt_rv=initial_log_rt_rv,
baseline_rt_process=DifferencedAR1(autoreg=0.9, innovation_sd=0.05),
subpop_rt_deviation_process=subpop_rt_deviation_process,
n_initialization_points=len(gen_int_pmf),
)
def make_sampler(m):
return lambda: m.sample(
n_days_post_init=n_days,
subpop_fractions=subpop_fractions,
)
deviation_samples[name] = Predictive(
make_sampler(model), num_samples=n_samples
)(random.PRNGKey(42))
Code
# Plot deviations for first subpopulation
dev_data = []
for name in subpop_configs.keys():
devs = np.array(
deviation_samples[name]["latent_infections::subpop_deviations"]
)[:, n_init:, 0]
for i in range(n_samples):
for d in range(n_days):
dev_data.append(
{
"day": d,
"deviation": float(devs[i, d]),
"sample": i,
"process": name,
}
)
dev_df = pd.DataFrame(dev_data)
Code
(
p9.ggplot(dev_df, p9.aes(x="day", y="deviation", group="sample"))
+ p9.geom_line(alpha=0.15, size=0.4, color="purple")
+ p9.geom_hline(yintercept=0, color="black", linetype="dotted")
+ p9.facet_wrap("~ process", ncol=2)
+ p9.scale_x_continuous(expand=(0, 0))
+ p9.labs(
x="Days",
y="Deviation δ₁(t)",
title="Subpopulation Deviation Trajectories (first subpop)",
subtitle=f"{n_samples} prior samples; baseline uses DifferencedAR1",
)
+ theme_tutorial
)
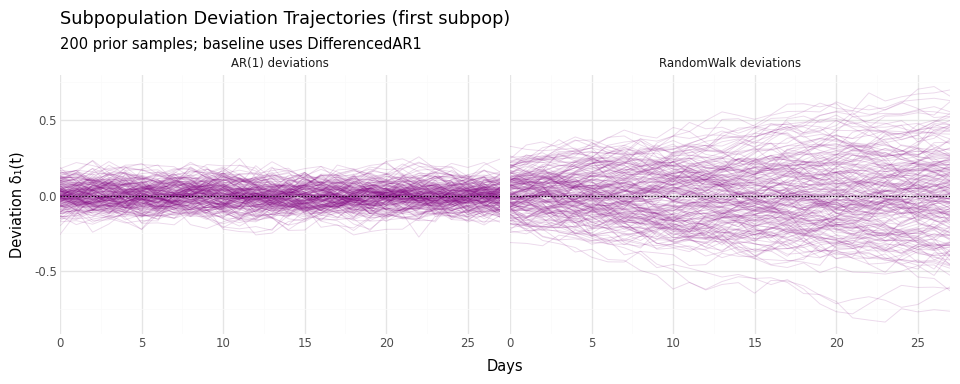
Figure 6: Subpopulation deviations under different temporal processes. AR(1) deviations decay toward zero; RandomWalk deviations can diverge.
Interpretation:
- AR(1) deviations stay close to zero—subpopulations track the jurisdiction average
- RandomWalk deviations can grow—subpopulations can diverge substantially
Choose based on your scientific question: Do you expect local differences to persist or decay?
Tuning Hyperparameters
The autoreg and innovation_sd parameters control how constrained the
prior is. Let’s compare a “weakly informative” vs “more informative”
configuration:
Code
configs = {
"Weakly informative\n(autoreg=0.9, sd=0.05)": {
"baseline": DifferencedAR1(autoreg=0.9, innovation_sd=0.05),
"subpop": AR1(autoreg=0.8, innovation_sd=0.05),
},
"More informative\n(autoreg=0.7, sd=0.02)": {
"baseline": DifferencedAR1(autoreg=0.7, innovation_sd=0.02),
"subpop": AR1(autoreg=0.6, innovation_sd=0.02),
},
}
tuning_samples = {}
for name, cfg in configs.items():
model = HierarchicalInfections(
gen_int_rv=gen_int_rv,
I0_rv=I0_rv,
initial_log_rt_rv=initial_log_rt_rv,
baseline_rt_process=cfg["baseline"],
subpop_rt_deviation_process=cfg["subpop"],
n_initialization_points=len(gen_int_pmf),
)
def make_sampler(m):
return lambda: m.sample(
n_days_post_init=n_days,
subpop_fractions=subpop_fractions,
)
tuning_samples[name] = Predictive(
make_sampler(model), num_samples=n_samples
)(random.PRNGKey(42))
Code
# Build comparison dataframe
tuning_data = []
for name, samples in tuning_samples.items():
rt = np.array(samples["latent_infections::rt_baseline"])[:, n_init:, 0]
for i in range(n_samples):
for d in range(n_days):
tuning_data.append(
{
"day": d,
"rt": float(rt[i, d]),
"sample": i,
"config": name,
}
)
tuning_df = pd.DataFrame(tuning_data)
Code
(
p9.ggplot(tuning_df, p9.aes(x="day", y="rt", group="sample"))
+ p9.geom_line(alpha=0.1, size=0.4, color="steelblue")
+ p9.geom_hline(yintercept=1.0, color="red", linetype="dashed", alpha=0.7)
+ p9.facet_wrap("~ config", ncol=2)
+ p9.scale_x_continuous(expand=(0, 0))
+ p9.coord_cartesian(ylim=(0, 3.0))
+ p9.labs(
x="Days",
y="Rt (baseline)",
title="Effect of Hyperparameter Tuning",
)
+ theme_tutorial
)
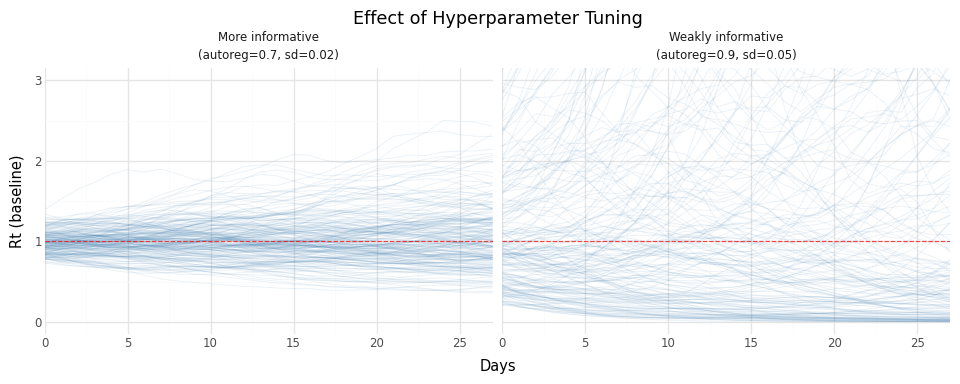
Figure 7: Effect of hyperparameter choices on prior Rt distribution. Tighter parameters produce more constrained trajectories.
Guidance:
- Lower
autoreg→ faster reversion → tighter distribution - Lower
innovation_sd→ smaller daily changes → smoother trajectories - With sparse data, more informative priors prevent explosive estimates
- With abundant data, weakly informative priors let the data speak
Verifying Model Structure
Before using HierarchicalInfections in a full model, verify the
hierarchical structure is working correctly:
Code
# Check that deviations sum to zero (identifiability constraint)
with numpyro.handlers.seed(rng_seed=123):
with numpyro.handlers.trace() as trace:
latent_infections.sample(
n_days_post_init=n_days,
subpop_fractions=subpop_fractions,
)
deviations = trace["latent_infections::subpop_deviations"]["value"]
deviation_sums = jnp.sum(deviations, axis=1)
print(
f"Max |sum of deviations|: {float(jnp.max(jnp.abs(deviation_sums))):.2e}"
)
print("(Should be ~0 due to sum-to-zero constraint)")
Max |sum of deviations|: 2.98e-08
(Should be ~0 due to sum-to-zero constraint)
Code
# Check that aggregate = weighted sum of subpopulations
with numpyro.handlers.seed(rng_seed=123):
inf_agg, inf_all = latent_infections.sample(
n_days_post_init=n_days,
subpop_fractions=subpop_fractions,
)
computed_agg = jnp.sum(inf_all * subpop_fractions[None, :], axis=1)
max_diff = float(jnp.max(jnp.abs(computed_agg - inf_agg)))
print(f"Max difference (aggregate vs weighted sum): {max_diff:.2e}")
print(
"Hierarchical structure verified!"
if max_diff < 1e-5
else "ERROR: Mismatch detected"
)
Max difference (aggregate vs weighted sum): 0.00e+00
Hierarchical structure verified!
Summary
What We Covered
-
HierarchicalInfectionsmodels subpopulation structure with shared baseline Rt and local deviations -
Model inputs each affect the dynamics differently:
- Generation interval: Determines response speed (fixed by pathogen biology)
- I0: Scales the entire infection trajectory
- initial_log_rt: Sets initial growth/decline direction
- Temporal processes: Control how Rt evolves over time
-
All inputs are RandomVariables: In inference, use
DistributionalVariablewith priors for quantities you want to estimate -
Prior predictive checks reveal model beliefs before seeing data—critical when data is sparse or noisy
Decision Guide
| Input | Consideration |
|---|---|
| Generation interval | Fixed by pathogen; shorter = faster dynamics |
| I0 | Often poorly identified; prior matters |
| initial_log_rt | Sets starting direction; can often be estimated |
| Temporal process | AR1 for regularization; RandomWalk for flexibility |
autoreg, innovation_sd |
Lower values = tighter prior; adjust if trajectories explode |
Code Summary
from pyrenew.latent import HierarchicalInfections, AR1, DifferencedAR1
from pyrenew.deterministic import DeterministicPMF, DeterministicVariable
# Configure the latent process
latent = HierarchicalInfections(
gen_int_rv=DeterministicPMF("gen_int", gen_int_pmf),
I0_rv=DeterministicVariable("I0", 0.001),
initial_log_rt_rv=DeterministicVariable("initial_log_rt", 0.0),
baseline_rt_process=DifferencedAR1(autoreg=0.9, innovation_sd=0.05),
subpop_rt_deviation_process=AR1(autoreg=0.8, innovation_sd=0.05),
n_initialization_points=len(gen_int_pmf),
)
# Sample (population structure provided at sample time)
infections_agg, infections_all = latent.sample(
n_days_post_init=28,
subpop_fractions=subpop_fractions,
)
When building the full multisignal model, you would instantiate a
PyrenewBuilder object and call the configure_latent method to
specify the latent process model structure: generation interval, initial
infections, and temporal dynamics. See tutorial: